Zespół Klippla-Trenaunaya
NORD Member Organizations
NORD z wdzięcznością dziękuje Johnowi B. Mullikenowi, MD, współdyrektorowi Centrum Anomalii Naczyniowych; Dyrektorowi Craniofacial Center, Boston Children's Hospital, za pomoc w przygotowaniu niniejszego raportu.
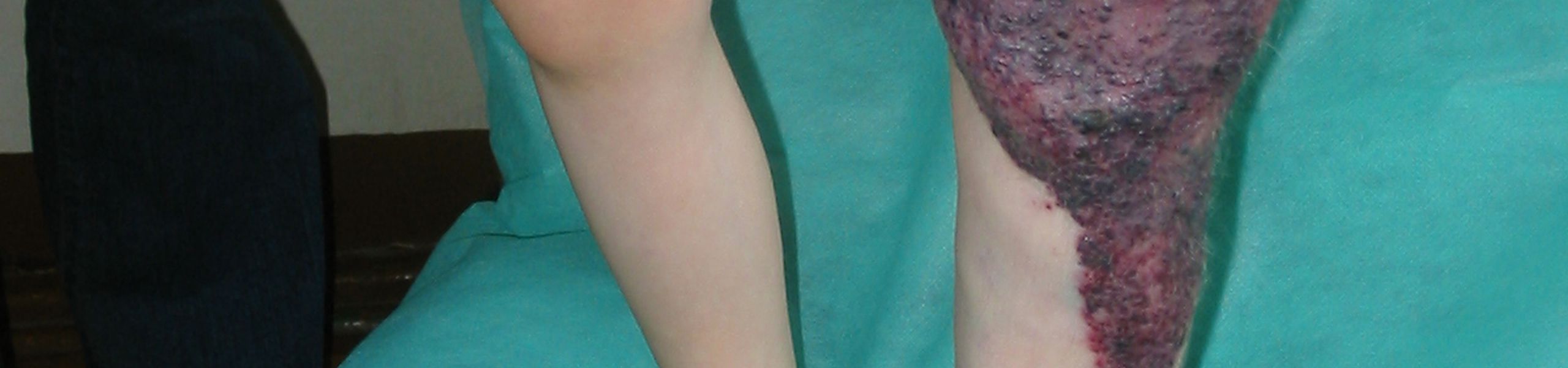
Zespół Klippel-Trenaunay (KTS) jest rzadkim zaburzeniem, które występuje przy urodzeniu (wrodzony) i charakteryzuje się triadą skórnych wad rozwojowych naczyń włosowatych („plama z wina porto”), anomaliami limfatycznymi i nieprawidłowymi żyłami w połączeniu ze zmiennym przerostem tkanki miękkiej i kości. KTS występuje najczęściej w kończynie dolnej, rzadziej w kończynie górnej i tułowiu.
Wprowadzenie
Eponim KTS wzbudził kontrowersje w literaturze medycznej od pierwszego doniesienia o chorobie na początku XX wieku. Francuscy lekarze Klippel i Trenaunay opisali pacjentów z przebarwieniami naczyń włosowatych (niesłusznie wówczas nazywanymi „naczyniakami krwionośnymi”), żylakami żylnymi i przerostem. Mniej więcej w tym samym czasie angielski dermatolog Parkes Weber zgłosił połączenie „naczyniaków krwionośnych” i przerostu kończyny. Przez wiele lat nazwiska wszystkich trzech lekarzy były łączone jako mylące (i niepoprawne) określenie „zespół Klippla-Webera-Trenaunaya”, które nadal (niestety) jest czasami używane do dziś.
Od drugiej połowy XX wieku powszechnie wiadomo, że zespoły Parkesa Webera i Klippel-Trenaunay są zupełnie inne. Zespół Parkesa Webera składa się z wielu mikroskopijnych połączeń tętniczo-żylnych o szybkim przepływie ze zmiennym wybarwieniem naczyń włosowatych powiększonej kończyny (zwykle kończyny dolnej). W badaniach genetycznych wielu z tych pacjentów ma dominującą mutację linii zarodkowej w genie RASA1. W przeciwieństwie do KTS jest połączonym zaburzeniem naczyniowym o powolnym przepływie, obejmującym nieprawidłowe naczynia włosowate (C), układ limfatyczny (L) i żyły (V). Dlatego wielu badaczy używa skrótu CLVM zamiast KTS i ogranicza oznaczenie pacjentów, którzy mają wszystkie trzy anomalie naczyniowe. Inni autorzy szerzej stosują termin KTS i obejmują pacjentów tylko z przebarwieniami naczyń włosowatych (CM) lub tylko z anomaliami naczyń włosowatych i żylnych (CVM) w kończynie, przy braku zaburzeń limfatycznych. Po zidentyfikowaniu mutacji linii germinalnej i somatycznej możliwe będzie dokładniejsze określenie pacjentów z różnymi kombinacjami anomalii naczyniowych.
Znaki i objawy
Malformacja naczyń włosowatych (CM)
Po urodzeniu KTS przedstawia rozproszone, geograficzne plamy kapilarne. Z wiekiem powierzchnia CM zostaje usiana maleńkimi pęcherzykami limfatycznymi, które często wydzielają klarowny płyn i stają się czarne z powodu krwawienia wewnątrz zmian chorobowych.
Wady układu limfatycznego (LM)
LM objawia się miejscowym lub uogólnionym przerostem spowodowanym anomaliami mikro- i makrocystycznymi, czasami w połączeniu z obrzękiem limfatycznym. Często występuje obrzęk limfatyczny i odkładanie się tłuszczu na przeciwległej stopie. Anomalie limfatyczne mogą również wystąpić w miednicy, pęcherzu i dolnym odcinku przewodu pokarmowego. Często występują torbiele limfatyczne w śledzionie. LM jest udokumentowane za pomocą ultrasonografii i / lub MRI. Limfografia pokazuje, że obrzęk limfatyczny jest wynikiem zmniejszonej liczby lub braku kanałów limfatycznych.
Infekcje epizodyczne (zapalenie tkanki łącznej) są częste i prawdopodobnie związane ze słabym drenażem limfatycznym kończyny.
Malformacja żylna (VM)
Nieprawidłowości żylne są zawsze obecne, ale zmienne i dotyczą całej chorej kończyny. Zwykle występują anomalne żyły embrionalne zwane „układem brzeżnym”. Rozszerzanie żył powierzchownych może nie być widoczne w okresie niemowlęcym, ale staje się bardziej widoczne wraz z wiekiem. LM i VM mogą również obejmować narządy miednicy lub jamy brzusznej, powodując krwawienie z odbytu, pochwy lub pęcherza moczowego. Nieprawidłowe złogi tłuszczowe towarzyszą anomaliom żylnym i limfatycznym.
U wielu pacjentów z rozległymi nieprawidłowymi żyłami występuje stan hematologiczny o niskim stopniu złośliwości, zwany „miejscową koagulopatią wewnątrznaczyniową” (ang. Localized intravascular coagulopathy) (LIC), którą można określić, mierząc podwyższone D-dimery we krwi. Stojąca krew w rozszerzonych żyłach może krzepnąć i wywołać uogólnione zaburzenie krwawienia zwane „rozsianą koagulopatią wewnątrznaczyniową” (DIC).
Przerost
Powiększenie kończyny może być minimalne lub groteskowe. Przerost długości jest typowy; jednak u niektórych pacjentów kończyna dotknięta chorobą jest krótsza niż normalnie. Często dochodzi do powiększenia przeciwnej stopy.
Przyczyny
Przyczyną KTS jest mutacja w prymitywnych komórkach tworzących kończynę, która ma stać się naczyniami krwionośnymi i limfatycznymi, tłuszczem i kościami. Jest to mutacja somatyczna, co oznacza, że występuje po zapłodnieniu w genie PIK3CA. Ta zmiana genetyczna nie występuje w komórkach rozrodczych (komórkach jajowych i nasieniu), dlatego KTS nie może być przenoszony w rodzinie.
Dotknięte populacje
Zespół Klippel-Trenaunay jest rzadkim zaburzeniem dotykającym w równej liczbie mężczyzn i kobiety. Zaburzenie występuje na całym świecie.
Powiązane zaburzenia
KTS można pomylić z innymi złożonymi zaburzeniami naczyniowymi.
Zespół Parkesa Webera
Małe przecieki tętniczo-żylne w udzie
Żródło: https://rarediseases.org/rare-diseases/klippel-trenaunay-syndrome/